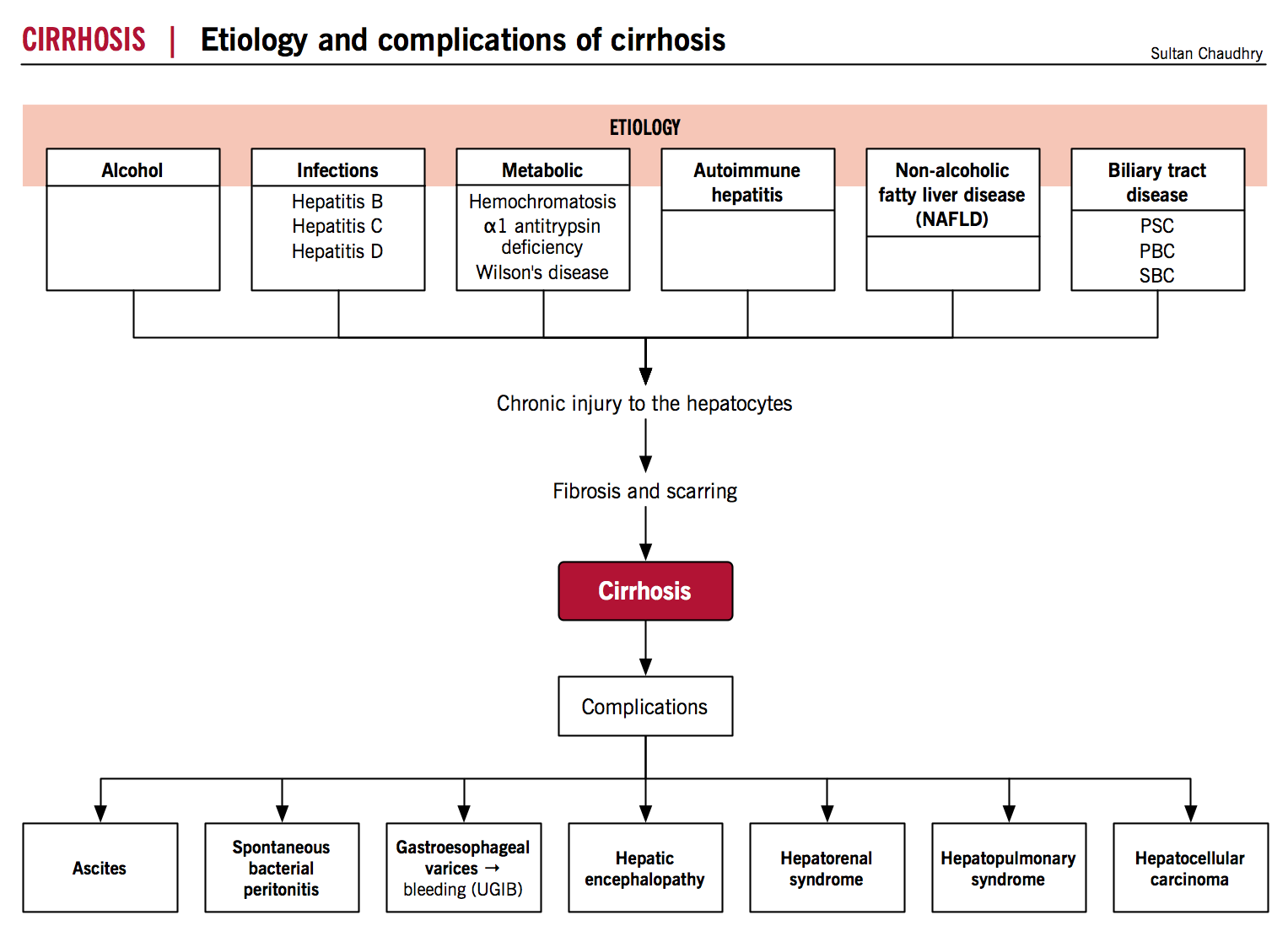
Definition
Annu Rev Pharmacol Toxicol 2005;45:605-28
Lancet 2008;371:838-51
- Acute liver injury leads to complex inflammatory reactions and matrix remodeling processes which result in the restoration of normal architecture
- Chronic liver injury leads to prolonged and dysregulated healing with accumulation of matrix proteins which ultimately progresses to cirrhosis
- Cirrhosis is an advanced stage of liver fibrosis characterized by
- Distortion of hepatic architecture associated with vascularized fibrotic septa surrounding islands of regenerating hepatocyte nodules
- Development of intrahepatic porto-hepatic and arterio-venous shunts within the fibrotic septa
- Major clinical consequences of cirrhosis are
- Impaired hepatocyte function
- Increased intrahepatic resistance (portal hypertension)
- Development of hepatocellular carcinoma (HCC)
- Traditionally cirrhosis is seen as an irreversible process which ultimately results in death but recent data suggest that regression or even reversal may be possible
Normal liver functions

Etiology
Patterns of hepatic injury
- Liver injury may be the result of infectious, metabolic, autoimmune, vascular, hereditary, or chemical factors
- The liver has a limited number of cellular and tissue responses to injury
- Hepatocyte degeneration and intracellular accumulations, hepatocyte necrosis and apoptosis, inflammation, regeneration and ultimately fibrosis
- Common etiologies include:
- Metabolic
- Non-alcoholic Fatty Liver Disease (NAFLD)/Nonalcoholic Steatohepatitis (NASH)
- Drugs and Toxins
- Alcoholic liver disease
- Drug-induced liver damage
- Infectious
- Viral hepatitis
- Autoimmune/Biliary disease
- Primary sclerosing cholangitis (PSC)
- Primary biliary cirrhosis (PBC)
- Autoimmune hepatitis
- Genetic and Infiltrative Disease
- Wilson’s disease
- Hemochromatosis
- alpha-1 antitrypsin deficiency
- Cryptogenic (~10-15%)
- Metabolic
NAFLD/NASH
- CMAJ 2005;172(7):899-905
- Clin Liver Dis 2007;11:75–104
- Primary NAFLD is considered the hepatic manifestation of the metabolic syndrome
- Multiple-hit process, initially involving insulin resistance and obesity, progresses to NASH due to increased inflammation, oxidative stress, apoptosis, and adipokine dysfunction
- Insulin resistance
- Insulin resistance enhances triglyceride (TG) lipolysis, inhibits esterification of free fatty acids (FFA) within the adipose tissue and inhibits FFA oxidation
- These increased levels of serum free fatty acids are then taken up by the liver and drive triglyceride production and hepatic steatosis
- Chronic hyperinsulinemia promotes de novo hepatic lipogenesis and activates profibrotic cytokines
- Hepatic lipid metabolism
- Lipids normally exported in very low density lipoproteins (VLDL) which are formed by microsomal triglyceride transfer protein (MTP) incorporating TG into apolipoprotein B (apo B)
- A reduction in MTP activity and apo B synthesis and secretion impairs hepatic lipid export and favours hepatic triglyceride accumulation
- Inflammatory and fibrotic mediators in NAFLD
- Adipocytokines (TNF alpha, leptin and adiponectin), FFA, mitochonddrial dysfunction, bacterial endotoxin and vascular disturbance are all implicated in hepatic inflammation and fibrosis through direct hepatotoxicity, generation of radical oxygen species (ROS) with subsequent lipid peroxidation, cytokine induction and liver damage
Alcoholic fatty liver
Alcohol 2004;34:9-19
- Heavy alcohol consumption may result in cirrhosis in 1 to 2 years or may be manifested several years after cessation of drinking
- Pint years can be used to measure alcohol damage, with 15 pint years being a reliable measure for cirrhosis (1 pint of whiskey = 16 oz = ~473 mL per day for 15 years)
- Several major factors play a role in the development of alcoholic fatty liver.
- Malnutrition
- Alcohol provides more calories per gram than carbs or proteins
- On average, ethanol provides about half of an alcoholic’s caloric intake leading to malnutrition
- Nutritional deficiencies can directly result in liver damage
- Alcohol promotes the degradation of nutrients
- Other ethanol-related complications (pancreatic insufficiency, impaired hepatic metabolism of nutrients, chronic gastric and intestinal mucosal damage) lead to malabsorption of ingested food
- Toxic effects of hepatic alcohol oxidation
- Oxidation of ethanol through alcohol dehydrogenase pathway produces acetaldehyde which is converted to acetate
- Acetaldehyde is directly toxic to the liver: it inhibits the repair of alkylated nucleoproteins, decreases activity of key enzymes, impairs mitochondrial oxygen use which can promote hypoxic/ischemic injury, depletes glutathione and increases toxic effects of free radicals
- Both reactions reduce NAD to NADH → excess NADH inhibits both the Krebs cycle and fatty acid oxidation with subsequent steatosis and hyperlipidemia
- Microsomal ethanol oxidizing system and oxidative stress
- Four-to-tenfold increase in activity of CYP 2E1 (contributes to the metabolic tolerance to ethanol which develops in the alcoholic)
- Induced activity results in oxidative stress which leads to ROS, lipid peroxidation, inflammation and mitochondrial damage which further worsens the oxidative stress within the cell
- Malnutrition
Drug-induced liver injury
Drug Safety 2007; 30 (4): 277-294
- Three different patterns of injury are recognized
- Hepatocellular
- The reactive metabolite of a drug undergoing metabolism in the hepatocyte induces cellular stress which leads to cell apoptosis or necrosis, triggering an immune response which itself increases sensitization leading to severe liver injury
- Cholestatic
- Canalicular excretion exposes bile duct cells to reactive metabolite resulting in injury to bile ductal cells and causing cholestasis
- Most drugs are associated with bile duct injury and inflammation
- Mixed
- Hepatocellular
Viral Hepatitis
Nat Clin Pract Gastr 2007;4(11):622-634
- Hepatitis A and E – usually nonfatal, self-limited disease characterized by a short period of disability followed by complete recovery. Acute liver failure is rare.
- Hepatitis B – a DNA virus (unlike other hepatotropic viruses which are RNA) may occur as a discrete entity or as co-infection with Hepatitis D (which requires hepatitis B presence). Hepatocellular carcinoma is a potential complication even in the absence of cirrhosis
- Hepatitis C – an RNA virus that may cause chronic infection in 80% of patients and cirrhosis in 15% of patients. The propensity to cirrhosis and liver cancer is increased in alcoholics
- Direct effect of HCV on hepatocytes
- HCV proteins modulate apoptosis and interact with hepatic lipid metabolism leading to inflammation, oxidative stress and steatosis
- Ultimately results in hepatic stellate cell activation, fibrosis and hepatocellular carcinoma
- Immune system mediated liver damage
- HCV disrupts both innate and adaptive immunity to establish persistent infection
- The immune system (NK and CD8 T lymphocytes in particular) tries to eradicate the virus and if unsuccessful, promotes hepatocyte damage and fibrosis through direct cellular toxicity and release of inflammatory cytokines
- Direct effect of HCV on hepatocytes
Primary biliary cirrhosis
Postgrad Med J 2008;84:23–33
- PBC is characterized by damage to and eventual loss of the biliary epithelial cells (BEC) lining small intrahepatic bile ducts as well as accompanying inflammation and granulomatous lymphocytic infiltration of the portal tract
- Unclear pathogenesis
- Immune mechanisms
-
- Characterised immunologically by the breakdown of immune self-tolerance to highly conserved mitochondrial and nuclear antigens
- IgA/IgM trancytosing across BEC inducing cellular dysfunction by interacting with peptides on mitochondria wall
- Antimitonchondrial antibody mediated cell toxicity
- PDC epitope derived cytotoxic T-cell response
- Actions of pro-inflammatory cytokines released by immune cells
-
- Non-immune mechanisms
-
- Oxidant stress secondary to macrophage activation
- Cytotoxic and pro-apoptotic effect of hypothetical environmental toxin in bile
- Cytopathic effect of postulated PBC-associated retrovirus
-
Primary sclerosing cholangitis
Digest Liver Dis 2010;42(390-400)
- PSC is a progressive disease characterized by inflammation, concentric fibrotic strictures and dilatation of the intra and/or extra hepatic bile ducts or gallbladder
- Important comorbidities
- Inflammatory bowel disease (~75% comorbidity, UC >> CD)
- Autoimmune hepatitis
- Cholangiocarcinoma
- Unclear pathogenesis
- Genetics
- Siblings of patients 9-39 times more likely vs general population
- Shared genetic risk with UC (9 times more likely to get UC independent of liver disease)
- Aberrant homing
- Abnormal expression of cellular adhesion molecule (MAdCAM-1) on the portal vein and sinusoidal endothelium is thought to recruit lymphocytes that subsequently cause biliary inflammation
- Autoimmunity
- Antibodies against biliary and colonic epithelium
- Hepatic T-cell tissue specific antigens recognized
- Perinuclear anti-neutrophil cytoplasmic antibody (pANCA) in up to 94% of patients
- Role of auto-antibodies of unknown significance
- Toxic bile
- Accumulation of bile beyond ability of hepatocyte to detoxify due to genetic alterations in bile acid transporters
- Leaky gut
- Innate immune response to bacterial products may initiate inflammation
- Toll like receptors (TLR-4 and 9) on biliary epithelial cells recognize bacterial products and viral DNA from bacterial colonization of biliary tree or intestinal mucosa
- Genetics
Autoimmune hepatitis
NEJM 2006;354:54-66
- Progressive inflammation of the liver characterized by the presence of circulating autoantibodies, hypergammaglobulinemia, fibrotic changes in the parenchyma and response to immunosuppressive therapy
- Unclear pathogenesis
- Postulated that an environmental agent triggers a cascade of T-Cell mediated events directed at hepatic antigens in a predisposed host leading to a progressive necroinflammatory and fibrotic process in the liver
- Type 1 or 2 based on circulating antibodies
- Putative autoantigens is the asiologlycoprotein receptor (liver specific membrane protein with high expression in periportal hepatocytes)
- Potential triggers for loss of self-tolerance
- Molecular mimicry (virual antigens; hepatitis, measles, cytomegalovirus, Epstein-Barr)
- Drugs (unclear if drugs unmask/induce autoimmune hepatitis or if there is a drug-induced hepatitis with accompanying autoimmune features)
Wilson’s disease
Mayo Clin. Proc. 2003;78(9):1126-1138
- Wilson’s disease is a rare autosomal recessive disorder of copper metabolism
- Caused by mutation of ATP7B gene resulting in impaired copper excretion into bile and inability to incorporate copper into ceruloplasmin
- Characterised by accumulation of copper in the liver and other organs (CNS and kidneys)
- Copper intake
- Average daily uptake of copper from GI tract is 1.5 to 5 mg (most absorbed in stomach and duodenum)
- Excess copper intake induces production of a metallothionein in the enterocyte that captures copper and sheds via normal senescence
- Estimated total body copper is 50-150 mg
- Copper metabolism
- Copper is taken up by albumin and 90% is transported to the liver shortly after absorption
- In the hepatocyte copper is bound to a cytosolic protein and to specific copper enzymes as well as incorporated into apo-ceruloplasmin which is secreted into the blood
- This requires the action of an ATPase termed ATP7B located on the canalicular membrane
- Ceruloplasmin carries majority of plasma copper and is eventually degraded in the liver and the copper eliminated in the bile (only physiologic route of excretion)
- Deficiency in ATP7B protein results in
- Decreased copper transport into bile
- Impairs copper incorporation into cerloplasmin
- Inhibits ceruloplasmin secretion into blood
- Accumulation of copper and toxic liver injury through ROS via the Fenton reaction
Hemochromatosis
- Gastroenterology 2010;139:393–408
- N Engl J Med 2012;366:348-59
- Hemochromatosis is homozygous-recessive disorder characterized by toxic accumulation of iron in the parenchymal cells of the liver and other vital organs (heart and endocrine glands)
- Common triad of micronodular cirrhosis, diabetes mellitus and skin pigmentation
- Iron intake
- Homeostatic balance requires only 1-3 mg of absorped iron per day
- Total body iron pool ~2-6 grams, 0.5 grams stored in the liver
- Hemochromatosis leads to net iron accumulation of 0.5-1 gram per year (mainly in the liver)
- Disease usually manifested when >20 grams of stored iron accumulated
- Iron cycle
- Iron is reduced at the apical membrane of the duodenal enterocyte, actively transported via DMT1, stored mostly as ferritin and either exported to plasma via the basolateral ferroportin transporter
- Absorbed iron circulates bound to transferrin and is used primarily by erythroid precursors in the synthesis of heme
- Hepatocytes store iron as ferritin and are the principal site of production of the peptide hormone hepcidin
- Hepcidin blocks the release of iron from enterocytes and reticuloendothelial macrophages by degrading the iron exporter ferroportin thereby diminishing the ability of the cells to transfer iron to the plasma compartment
- Reticuloendothelial macrophages clear senescent erythrocytes and release the iron from heme to export it to the circulation or store it in ferritin
- These cells are the major hepcidin regulated iron repository, releasing up to 25 mg of iron per day
- Hemochromatosis can be caused by defects in any of the proteins that limit entry of iron into the blood leading to excessive iron accumulation
- Defects in hepcidin (most common) or any of the proteins that regulate it (HJV, TfR2, HFE) lead to an inability to adjust hepcidin expression to current iron needs leading to unrestricted flow of iron into the plasma compartment instead of being stored as ferritin
- Clinical features of the disease depend on which gene regulator is involved
- Defects in hepcidin (most common) or any of the proteins that regulate it (HJV, TfR2, HFE) lead to an inability to adjust hepcidin expression to current iron needs leading to unrestricted flow of iron into the plasma compartment instead of being stored as ferritin
- Excessive iron is directly toxic to cells
- Lipid peroxidation via iron-catalyzed free radical reactions
- Stimulation of collagen formation by activation of hepatic stellate cells
- Interaction of reactive oxygen species and iron with DNA leading to cell injury and predisposition to hepatocellular carcinoma
Alpha-1 antitrypsin deficiency
Curr Gastroenterol Rep 2006;8:14–20
- Alpha-1 antitrypsin deficiency is an autosomal recessive disease affecting the lung and liver
- Alpha-1 antitrypsin molecule is a serine protease inhibitor predominantly produced in the liver (second only to albumin in daily synthesis and serum predominance)
- Most important physiologic functions are the protection of pulmonary tissue from aggressive proteolytic enzymes (especially neutrophil elastase) and regulation of pulmonary immune processes.
- Encoded by the SERPINA1 gene (also known as PI)
- Highly pleomorphic with extensive allelic variation
- Disease manifestation associated with null variants or genotypes resulting in impaired gene expression, translation or protein synthesis
- Majority of individuals with lung or liver disease are homozygous for alleles Z or S or heterozygotes with one wild type and one S/Z allele
- These null variants cause abnormalities in the tertiary structure of molecule
- Protein synthesis in the rough endoplasmic reticulum (ER) of the hepatocyte is delayed
- Approximately 85% of molecules polymerize into large conglomerates
- Quality control systems recognize most mutant molecules as abnormal and shunt to proteolytic mechanisms leading to few non-polymerized molecules released into blood
- Antiproteolytic activity against neutrophil elastase reduced to 15-20% of normal serum levels leading to pulmonary damage
- Those polymers that cannot be further processed accumulate and form inclusions in the ER
- Triggers activation of variety of cellular responses
- Autophagy upregulation
- Mitochondrial damage
- Activation of apoptotic pathways
- Activation of ER stress pathways
- Eventually results in hepatocyte injury and death
- Increased risk of liver fibrosis and cirrhosis
- Amount of polymerized molecules correlate with stage of cirrhosis
- Likely that important environmental and genetic disease modifiers (as yet unknown) affect rate and magnitude of hepatocellular death
Pathogenesis
- Annu Rev Pharmacol Toxicol 2005;45:605-28
- Clin Liv Dis 2009;12:915-937
- Eur J Gastroenterol Hepatol 2010;23:8-22
- Lancet 2003;362:1819-27
- Lancet 2008;371:838-51
- Med Clin N Am 2009;93:787-799
- NEJM 2008;358:2378-87
- Gastroenterology 2008;134:1641–1654
- N Engl J Med. 2009 Sep 24;361(13):1279-90.
- Hepatic stellate cells (HSC) located in the space of Disse comprise 5-10% of hepatic cells
- Parenchymal injury (acute or chronic) -> apoptosis and/or necrosis of hepatocytes -> phagocytosis of apoptotic cells and inflammation due to necrosis leads to activation of Kuppfer cells (sinusoidal macrophages), endothelial cells, platelets and leukocytes
- Leukocytes lead to generation of lipid peroxides, reactive oxygen species and a number of cytokines (TGF beta, IL-1, TGF alpha, HGF, PDGF, and EGF)
- Stimulates regeneration of adjacent hepatocytes (HGF) ultimately contributing to nodule formation
- Promote induction/expression of variety of cytokines/chemokines and receptors on neighbouring quiescent hepatic cells and activates them
- Remodeling of matrix also leads to activation of nearby HSC
The role of the activated hepatic stellate cell (AHSC) and myofibroblasts.
- Acquisition of the myofibroblast phenotype
- Fibroblasts of portal, perivascular, and periductular tissues transform into myofibroblasts
- HSC change phenotype from a quiescent vitamin A-rich phenotype to a myofibroblastic phenotype (AHSC)
- Though derived from two distinct cell populations, the phenotypic and functional properties of both types of myofibroblasts are grossly similar
- AHSC show de novo fibrogenic properties
- Proliferation and accumulation of AHSC in areas of parenchymal necrosis due to
- High mitogenic capacity [stimulated by growth factors, vasoconstrictors (angiotensin II), metalloproteinases, adhesion molecules, intracellular pathways]
- Enhanced ability to escape apoptosis (TNF alpha and TGF beta, matrix components)
- Chemotaxis to site of injury via chemokines and growth factors
- Secretion of proinflammatory cytokines and chemokines
- Synthesis of a large panel of matrix proteins and inhibitors of matrix degradation
- Produce metalloproteinases (MMP) and MMP activators as well as tissue inhibitors of MMPs (TIMPS)
- Early on -> express MMP but not TIMP -> normal matrix is degraded and replaced with fibrillar collagen
- Later on -> do not express MMP but express TIMP -> dramatic reduction in collagenolytic activity within liver
- Eventually lead to progressive fibrosis and scar formation
- Proliferation and accumulation of AHSC in areas of parenchymal necrosis due to
Fibrosis
- Fibrosis is the net deposition of excess extracellular matrix (ECM) arising from an imbalance between hepatic fibrogenesis and fibrinolysis leading to replacement of injured tissue by a collagenous scar
- Liver fibrosis results from the perpetuation of the normal wound healing response resulting in an abnormal continuation of fibrogenesis (connective tissue production and deposition)
- Fibrosis progresses at variable rates depending on the cause of liver disease, environmental factors, and host factors
- There is a quantitative change in expression and maintenance of different types of collagen
- Normally type I and III collagen found around portal tracts and central veins, type IV collagen in space of Disse
- In cirrhosis the balance shifts to a predominance of type I and III collagen (fibril forming) within the space of Disse -> these types cross-link into collagen bundles which build up a network resistant to fibrolysis
- Leads to a remodeling to a fibrotic matrix
- The fenestrated endothelia that line the hepatic sinusoids are lost (sinusoidal capillarization) due to fibrosis in the space of Disse which increases intrahepatic resistance and contributes to portal hypertension
- The AHSC shift balance to fibrosis by synthesizing fibrotic matrix proteins and components that inhibit fibrosis degradation
- The resultant vascular distortion leads to an alteration of the relationship between vascular structures which compromises exchange between hepatic sinusoids and adjacent parenchyma
- There is obliteration of portal and/or hepatic veins due partly to intraluminal thrombosis, intimal thickening and inflammation which leads to hypoxia, then ischemia and finally parenchymal extinction (loss of liver cell plates and accompanying sinusoids)
- Subsequent angiogenesis contributes to the arterialization at the level of portal tracts with increased formation of de novo intra-hepatic arterio-venous shunts which effectively bypass the parenchyma
- Blood flows preferentially through these vascular channels due to the relative low resistance of these shunts and leaves the remainder of the hepatic parenchyma almost bereft of meaningful blood flow which leads to hepatocyte dysfunction
Pathophysiology
- Lancet 2008;371:838-51
- Med Clin N Am 2009;93:787-799
Clinical features
| Symptom/sign | Description | Cause/mechanism |
| Jaundice and scleral icterus | Yellow discoloration of skin, cornea, and mucous membranes | Increasing serum bilirubin due to compromised hepatocyte excretory function |
| Spider angiomata | Central arteriole with tiny radiating vessels, mainly on trunk and face | Raised estradiol (decreased estradiol degradation in liver) |
| Palmar erythema | Erythema sparing central portion of palm | |
| Gynecomastia, loss of male hair pattern | Benign proliferation of glandular male breast tissue | Elevated estradiol levels |
| Hypogonadism | Mainly in alcoholic cirrhosis and hemochromotasis | Raised estradiol (decreased estradiol degradation in liver)
Direct toxic effect of alcohol or iron |
| Nodular liver | Irregular, hard surface on palpation | Fibrosis, irregular regeneration
Hepatocellular carcinoma |
| Splenomegaly | Enlarged on palpation or in ultrasound | Portal hypertension (see below for description)
Splenic congestion |
| Caput medusae | Prominent veins radiating from umbilicus | Portal hypertension
Reopening of umbilical vein that shunts blood from portal vein |
| Ascites | Proteinaceous fluid in abdominal cavity (clinically detected when ≥ 1.5L) | Portal hypertension
Hypoalbuminemia – loss of oncotic pressure |
| Petechiae, purpura, bleeding | Thrombocytopenia, gastric varices, hemorrhoids | Thrombocytopenia – sequestration in the spleen as well as loss of thrombopoietin production (produced by the liver)
Portal hypertension Decreased clotting factors – damaged liver fails to produce adequate levels of coagulation factors. |
| Leukonychia/Terry’s nails | Horizontal white bands or proximal white nail plate | Hypoalbuminemia
Decrease in vascularity and an increase in connective tissue in the nail bed |
| Finger clubbing | Loss of normal angle between nailbed and fold | Hypoxia associated with hepatopulmonary syndrome or portopulmonary hypertension
Clear mechanisms unknown. |
| Dyspnea, hypoxia | Dyspnea and arterial deoxygenation greater in upright position vs recumbent position | Hepatopulmonary syndrome (see below for more details) |
| Anorexia, fatigue, weight loss, muscle wasting | Occurs in >50% of patients with cirrhosis | Catabolic metabolism by diseased liver
Secondary to anorexia Hepatocellular carcinoma Anemia |
| Dupuytren’s contracture | Fibrosis and contraction of palmar fascia | Enhanced oxidative stress, increased inosine (alcohol exposure or diabetes) |
| Asterixis | Asynchronous flapping motion of dorsiflexed hands | Dysregulation of ammonia metabolism/hepatic encephalopathy
Disinhibition of motor neurons |
| Confusion, hepatic encephalopathy | Altered level of consciousness due to liver failure | Dysregulation of ammonia metabolism – ammonia is taken up and excreted in the bile by the hepatocytes.
Alteration of the blood brain barrier Disruption in neurotransmission Alterations in GABA pathways Spontaneous bacterial peritonitis |
| Fetor hepaticus | Sweet, pungent smell | Volatile dimethylsulfide, especially in portosystemic shunting and liver failure |
| Type II diabetes mellitus | Occurs in 15-30% of patients with cirrhosis | Disturbed glucose use or decreased insulin removal by liver |
| Acute kidney Injury (oliguria, volume overload) | Drop in glomerular filtration rate (GFR) or plasma elevation of Creatinine. | Bleeding or loss of effective arterial circulating volume in cirrhosis patients
Use of diuretics and NSAIDs in cirrhosis – more susceptible to developing AKI due to the high dependency on prostaglandins to maintain GFR. Spontaneous Bacterial Peritonitis (SBP) Hepatorenal syndrome I and II |
Laboratory abnormalities
| Lab findings | Description | Cause/mechanism |
| AST/ALT | Normal or moderately raised | Leakage from damaged hepatocytes; AST/ALT ratio often ~2:1 in alcoholic cirrhosis (due to alcohol induced vitamin B6 deficiency which is a cofactor in the synthesis of both but ALT>AST) |
| ALP | Increased by less than threefold (other than in primary sclerosing cholangitis or biliary cirrhosis) | Cholestasis |
| GGT | More specific for liver than ALP, elevated in obstruction, high concentrations in active alcoholics | Cholestasis |
| Anemia | Microcytic, normocytic or macrocytic | Due to bleeding (esophageal varices)
Splenomegaly Malnutrition (folate/B12) Chronic disease Bone marrow suppression (alcohol) |
| Prothrombin time | Decreased in advanced cirrhosis | Decreased hepatic production of factor V/VII (while thrombin production is maintained), Vitamin K deficiency |
| Thrombocytopenia | Decreased platelets | Splenomegaly
Reduced hepatic production of thrombopoietin Direct toxic effect of alcohol on the bone marrow |
| Albumin | Decreased in advanced cirrhosis | Decreased hepatic production
Sequestration into ascites and interstitium Malnutrition |
| Immunoglobulins | Increased (mainly IgG) | Shunting of portal venous blood carrying (intestinal) antigens to lymph tissues with resultant stimulation of plasma cells |
| Hyponatremia | Sodium and water imbalance | Inability to excrete free water via kidneys due to increased activity of ADH
Arterial underfilling leading to RAAS activation and water retention. Secondary to complications of portal hypertension and splanchnic vasodilation |
Systemic syndromes
| Clinical entity | Cause/mechanism |
| Portal hypertension | Architectural and functional changes in liver resulting in increased resistance to portal flow leading to portosystemic collaterals, splanchnic vasodilation, expanded plasma volume and arterial underfilling |
| Portopulmonary hypertension | Vasoactive substances not filtered by the damaged liver causing pulmonary artery vasoconstriction, remodeling of vessels and in situ thrombosis resulting in increasing pulmonary vasculature resistance and ultimately right heart failure |
| Hepatopulmonary syndrome | Intrapulmonary vascular dilatation due to nitric oxide (NO) or arteriovenous communication (with a predominance at lung bases) causes ventilation/perfusion mismatch and/or shunting which worsens when upright due to dependent pooling of blood at bases |
| Hepatorenal syndrome | Renal failure caused by intense vasoconstriction of the renal circulation as a consequence of extreme underfilling of arterial circulation due to splanchnic vasodilation (due to high NO levels) and dysregulation of vasocoactive systems in the setting of advanced liver disease |