Definition
It is important to note that heart failure is not a diagnosis. Rather, it represents a constellation of signs and symptoms resulting from the inability of the heart to pump blood forward at a sufficient rate to meet the metabolic demands of the body (forward failure) or the ability to do so only if the cardiac filling pressures are abnormally high (backward failure), or both. Often, it is the final and most severe manifestation of almost any form of cardiac disease.
Extra-cardiac conditions resulting in inadequate tissue perfusion, such as severe hemorrhage, or increased metabolic demands, such as in hyperthyroidism, can also cause the above definition to be met, but will not be addressed here.
Etiology
Am J Cardiol. 2005 May 2;95(9A):8B-13B.
Lilly, Pathophysiology of Heart Disease, 2007
Predominantly systolic dysfunction is seen in a majority of patients, while others exhibit mainly diastolic dysfunction. Components of both can also be found.
Systolic dysfunction – diminished ability to eject blood due to:
- Impaired ventricular contractility: destruction or abnormal function of myocytes, fibrosis
- Increased afterload – increases resistance to flow
Diastolic dysfunction
- Impaired ventricular relaxation
- Impaired ventricular filling due to increased ventricular wall stiffness
This classification may help distinguish causes in terms of their impact on normal heart physiology, heart failure can also be thought of clinically as right- versus left-sided heart failure.
Systolic and diastolic dysfunction leading to left- and right-sided heart failure
| Left-sided heart failure | Right-sided heart failure | |
| Systolic dysfunction | ||
| Impaired ventricular contractility |
|
|
| Increased afterload |
|
|
| Diastolic dysfunction | ||
| Impaired ventricular relaxation |
|
|
| Impaired ventricular filling |
|
|
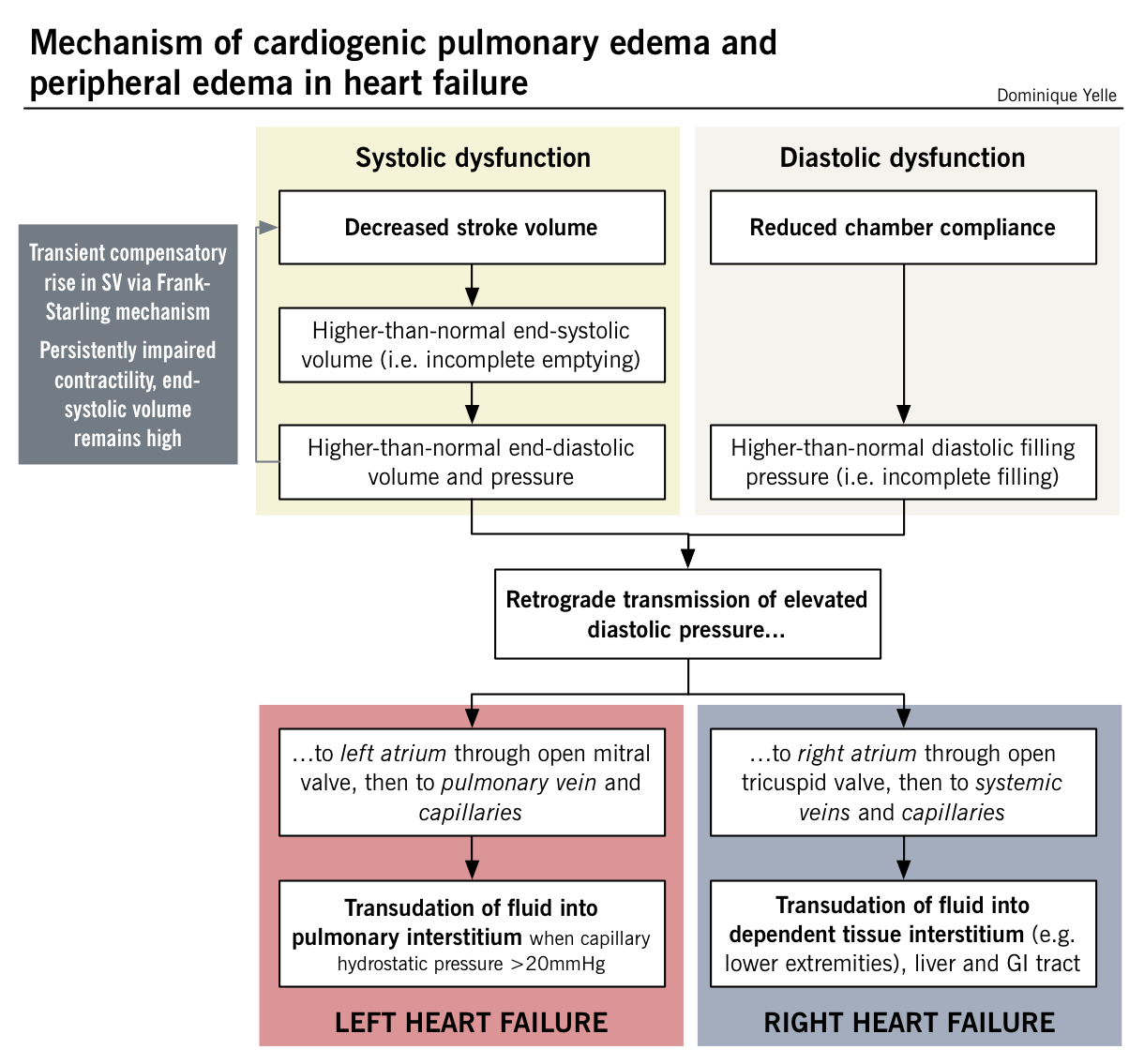
NOTE: In contrast to the left ventricle, the right ventricle is thin-walled and highly compliant, and therefore adapts well to a wide range of filling volumes when working against a low pulmonary vascular resistance. However, it is quite susceptible to failure when there is a sudden increase in afterload, such as in left-sided heart failure (retrograde pressure transmission to the pulmonary circulation). Consequently, the most common cause of right-sided heart failure is left-sided heart failure. Although less common, isolated right heart failure does arise as a result of primary lung pathology and in such cases is referred to as cor pulmonale.
Pathogenesis
Myocyte loss and/or dysfunction
In addition to global mechanical dysfunction in heart failure, another key player in this process may be dysfunction at a cellular level.
- Myocyte loss
- Necrosis – resulting from insults such as MI or exposure to cardiotoxic drugs
- Apoptosis (programmed cell death) from elevated catecholamines, angiotensin II, inflammatory cytokines, and mechanical strain from increased wall stress
- Changes activated in expression of contractile proteins, ion channels, enzymes, receptors and secondary messengers
- Reduced cellular ability to maintain calcium homeostasis
- Changes in handling of high-energy phosphates
Compensatory mechanisms
These mechanisms attempt to maintain sufficient blood pressure to perfuse vital organs by compensating for the decrease in cardiac output that occurs in heart failure.
Frank-Starling mechanism
- Frank-Starling relationship: Ventricular output increases in relation to preload, i.e. with a greater stretch of myocardial fibers (larger diastolic volume), there will be a greater force of contraction generated
- In heart failure, a decreased stroke volume results in reduced chamber emptying, with higher than normal diastolic volume
- This induces a greater stroke volume for the subsequent contraction to help empty the ventricle and preserve forward cardiac output
- However this mechanisms has limits, and at markedly elevated diastolic volumes, the stretch of myofibers becomes too great and suboptimal for generating a strong contraction
Myocardial hypertrophy
- Wall stress is often increased in heart failure due to either ventricular dilatation or the need to generate high systolic pressures to overcome excessive afterload
- Wall stress is estimated from LaPlace’s relationship, in which the wall stress (σ) is proportional to ventricular pressure (P) and ventricular chamber radius (r), and inversely proportional to ventricular wall thickness (h)
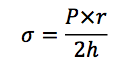
- In response to a sustained increase in pressure and chamber radius, hypertrophy of the ventricular myocytes is stimulated
- The increased mass of muscle fibers serves to maintain contractile force and counteract the elevated ventricular wall stress
- Eventually, the chamber may dilate out of proportion to wall thickness, resulting in excessive hemodynamic burden on the contractile units, rapid deterioration of ventricular function and worsening of symptomatology
- Lowering wall stress as a way to slow the remodelling process is a common therapeutic target
Neuro-hormonal mechanisms
- In the early stages of heart failure, these mechanisms help maintain a near normal perfusion to vital organs by increasing systemic vascular resistance as a way to balance the fall in cardiac output (blood pressure (BP) = cardiac output (CO) × total peripheral resistance (TPR)). In addition, activation of neuro-hormonal mechanisms leads to salt and water retention with a consequent increase in intravascular volume and preload, which maximizes stroke volume via the Frank-Starling mechanism.
- Renin-angiotensin-aldosterone system (RAAS): (See Nephrology for details of physiology). The pathway leads to the activation of angiotensin II.
- Angiotensin II
- Vasoconstriction: Increases TPR to maintain BP.
- Increased intravascular volume to increase preload to raise the SV via Frank-Starling mechanism. Angiotensin II does this by (i) stimulating thirst at hypothalamus and (ii) increasing aldosterone secretion at adrenal cortex.
- Aldosterone
- Increased water retention via increased sodium resorption. This increases preload, in turn increasing the SV
- Angiotensin II
- Antidiuretic hormone (ADH, aka vasopressin)
- Increased secretion thought to be induced by arterial baroreceptors (detecting decreased CO) and increased angiotensin II levels.
- Promotes water retention in the distal collecting tubule, in order to increase preload.
Adrenergic nervous system
- Decreased CO results in decreased perfusion pressure sensed by baroreceptors in carotid sinus and the aortic arch. Central and peripheral chemoreflex activation induces epinephrine, norepinephrine, and vasopressin release. This results in an increased sympathetic outflow to heart and peripheral circulation, and decreased parasympathetic tone.
- Increased HR and contractility directly increase cardiac output (CO = HR × SV)
- Peripheral vasoconstriction
- Venous: Increases preload (venous return).
- Arteriolar: Raises peripheral vascular resistance, to maintain BP.
Unfortunately, despite these compensatory mechanisms, there is progressive decline in the heart’s ability to contract and relax in the face of persistent hemodynamic challenges. Furthermore, chronic activation of the above mechanisms ultimately becomes maladaptive and induces further worsening of cardiac performance.
Deleterious consequences of compensatory mechanisms
- Continuous sympathetic activation results in downregulation of β-adrenergic receptors with decreased sensitivity to circulating catecholamines and less inotropic response.
- Increased heart rate augments metabolic demands and can further reduce performance by increasing myocardial cell death.
- Increased circulating volume and preload ultimately overwhelm Frank-Starling mechanism and heart’s ability to maintain forward flow, resulting in worsening of lung vasculature congestion.
- Increased total peripheral resistance results in higher afterload, impeding the left ventricle’s stroke volume and reducing cardiac output.
- Chronically elevated angiotensin II and aldosterone trigger production of cytokines, which activate macrophages an stimulate fibroblasts resulting in adverse heart remodelling.
Exacerbating factors in compensated heart failure
Heart failure can be clinically silent (i.e. asymptomatic) if compensatory mechanisms are sufficient to balance the degree of cardiac dysfunction, or alternatively if it is adequately managed medically. However, patients can become symptomatic, if decompensation occurs.
| Precipitants | Mechanism of exacerbation |
| Fever, infection, anemia, tachyarrhythmia, hyperthyroidism | Increased metabolic demands: Inability to sufficiently increase cardiac output to match disproportionately elevated demand of organ systems |
| Excessive salt content in diet, excessive fluid administration, renal failure | Increased circulating volume (preload): Promotes systemic and pulmonary congestion as the heart is unable to accommodate a larger preload (Frank-Starling mechanism overwhelmed) |
| Uncontrolled hypertension, pulmonary embolism | Increased afterload: Increased resistance against which ventricle must pump with consequent decrease in stroke volume |
| Negative inotropic medications (β-blockers, calcium channel blockers), ischemia | Impaired contractility: Reduced contractility impairs stroke volume and consequently decreases cardiac output |
| Bradyarrhythmia | Direct drop in cardiac output (CO = HR × SV) |
| Medical non-adherence | Multifactorial; e.g. if diuretics not taken adequately, can result in increased circulating volume (as above) |
Pathophysiology
| Signs and symptoms | Underlying mechanism |
| Left-sided heart failure | |
| Symptoms | |
| Shortness of breath | Increased pulmonary capillary oncotic pressure from left-sided backflow causes extravasation of fluid into the pulmonary interstitium, which then leads to reduced pulmonary compliance and increased airway resistance. There is also an increased ventilatory drive secondary to hypoxemia, a consequence of increased pulmonary capillary pressures and ventilation/perfusion mismatch due to inadequate CO. |
| Orthopnea | Redistribution of extravascular fluid from the periphery into dependent areas when supine (i.e. lungs) exacerbates dyspnea as the ventricles cannot adapt to the acute increase in volume; this results in increased pulmonary capillary pressure and worsening of interstitial pulmonary edema. |
| Paroxysmal nocturnal dyspnea | |
| Cough +/- frothy blood-tinged sputum | Caused by pulmonary congestion. Rupture of engorged bronchial veins can lead to hemoptysis. |
| Confusion and/or impaired memory | Manifestations of inadequate cerebral blood supply |
| Decreased urine output | Decreased renal perfusion during the daytime can sometimes lead to acute kidney injury and eventually renal failure. |
| Nocturia | At night when supine, blood flow is redistributed to the kidney, promoting perfusion and diuresis. |
| Signs | |
| Pulmonary crackles | Opening of small airways that were closed by interstitial edema prior to inspiration; initially present at lung bases where hydrostatic forces are greatest, but worsening pulmonary edema is associated with crackles in higher lung fields |
| Cardiac “asthma” | Coarse ronchi and wheezing caused by compression of conduction airways by pulmonary congestion |
| Accentuated P2 | Reflects increased pulmonary vascular pressures caused by elevated left-heart filling pressures |
| Mitral regurgitation murmur | May be auscultated if dilation of the left ventricle has excessively stretched the mitral valve annulus and spread the papillary muscles, preventing full closure of the mitral leaflets |
| Pulsus alternans | May present as a sign of advanced ventricular dysfunction |
| Right-sided heart failure | |
| Symptoms | |
| Peripheral edema, sacral edema, ascites and/or anasarca | Various manifestations of increased hydrostatic venous pressures |
| Hepatosplenomegaly (+/- right upper quadrant pain) | Portal and splenic circulation engorgement from inadequate right-sided forward flow |
| Anorexia/weight loss | Hepatic and intestinal congestion result in diminished appetite; there can also be occasional impaired intestinal fat absorption, and more rarely, protein-wasting enteropathy; total metabolism can also be increased secondary to increased myocardial oxygen consumption and excessive work of breathing |
| Signs | |
| Elevated JVP | Reflects elevated right-sided pressures and inadequate right-sided forward flow |
| Kussmaul sign | Paradoxical elevation of JVP with inspiration (as opposed to decrease) reflects increased right atrial pressure |
| Palpable right ventricular heave | Represents right ventricular enlargement and approximation of the chamber to the chest wall |
| Tricuspid regurgitation murmur | May be auscultated if dilation of the right ventricle has excessively stretched the tricuspid valve annulus and spread the papillary muscles, preventing full closure of the tricuspid leaflets |
| Right- or left-sided heart failure | |
| Symptoms | |
| Chest pain/pressure | Can occur as a result of primary myocardial ischemia from CAD or secondary to increased filling pressures, poor cardiac output (with consequent poor coronary diastolic filling) or hypoxemia |
| Palpitations | Can be secondary to sinus tachycardia due to decompensated heart failure, or atrial/ventricular tachyarrhythmias arising in dilated heart chambers |
| Chronic fatigue/weakness | Likely a result of the chronic state of hypoxemia from inadequate oxygen delivery to peripheral tissues, with generalized decreased muscle strength, decreased endurance and multi-organ system dysfunction |
| Cachexia | In part due to poor appetite, along with increased metabolic demands of increased work of breathing |
| Signs | |
| S3 gallop | Caused by abnormal filling of a dilated ventricle, often in systolic heart failure |
| S4 gallop | Results from forceful contraction of the atria into a stiffened ventricle; common in diastolic dysfunction |
| Cardiomegaly | Chronically increased workload and excessive volume cause ventricular dilatation and hypertrophy |
Treatment
Principles: The main goal of therapy is first and foremost to identify and aim to correct any underlying conditions causing heart failure. In an acute decompensation, the goal also includes eliminating acute precipitating causes that turned a compensating heart failure state into a decompensated state. Symptomatic management and modulation of neurohormonal mechanisms are also important therapeutic targets.
Chronic management
Non-pharmacologic management
Lifestyle modification:
- 10% weight loss
- Sodium < 2gm/day
- Fluid restriction (<2L/day)
- Smoking and drinking cessation
Pharmacologic management
- β-blockers: (Lancet. 1999 Jun 12;353(9169):2001-7, Lancet. 1999 Jan 2;353(9146):9-13. Lancet. 2003 Jul 5;362(9377):7-13)
- Counteract the harmful effects of sympathetic nervous system activation, may also assist in preventing tachyarrhythmias and myocardial ischemia
- Of all beta blockers, only metoprolol succinate (MERIT-HF trial), carvedilol (COMET trial), and bisoprolol (CIBIS-II trial) have demonstrated mortality benefit
- COMET – carvedilol superior to metoprolol reducing mortality in NYHA II+ & EF <35%
- CIBIS II – significant reduction in all-cause mortality and hospitalization in NYHA II+
- ACE inhibitors (ACEi) / angiotensin receptor blockers (ARBs): (N Engl J Med. 1992 Sep 3;327(10):685-91)
- Counteract the activated mediators of the RAAS to prevent their deleterious consequences and ultimately minimize cardiac remodelling
- Enalapril shown to have significant morbidity and mortality benefit (SOLVD & CONSENSUS trials)
- For patients unable to tolerate ACEi, both candesartan and valsartan have been studied and have been shown to have mortality benefit (CHARM & V-HeFT trials) – no added benefit to ACEi
- Hydralazine + nitrates (N Engl J Med. 2004; 351:2049-2057)
- Consider if patients unable to tolerate ACEi/ARBs or in African American patients with NYHA class III/IV
- A-HEFT trial – 40% reduction in mortality
- Aldosterone antagonists (N Engl J Med. 2011 Jan 6;364(1):11-21, N Engl J Med. 1999 Sep 2;341(10):709-17.)
- Aldosterone antagonist counteracts the harmful effects of aldosterone including salt retention, myocardial hypertrophy and potassium excretion
- Spironolactone: RALES trial – 34% mortality benefit in patients with heart failure NYHA Class III+ (patients already optimized on beta blocker and ACEi therapy)
- Epleronone: EMPHASIS-HF trial – 37% reduction in death from CV causes or hospitalization for HF NYHA Class II+
- Digoxin (N Engl J Med. 1997 Feb 20;336(8):525-33):
- Enhances cardiac contractility, but also blunts the compensatory sympathetic drive by increasing the sensitivity of baroreceptors, which decreases afterload
- No mortality benefit; only decreased hospitalizations (DIG trial)
- ICDs and cardiac resynchronization therapy (N Engl J Med. 2005 Jan 20;352 (3):225-37, N Engl J Med. 2009 Oct 1;361 (14):1329-38.):
- Pacemaker-based approach to treat patients with a wide QRS complex
- Purpose is to provide electromechanical coordination and improve ventricular synchrony in patients with severe systolic dysfunction and significant intraventricular conduction defects such as left bundle-branch block
- ICDs in patients with NYHA II/III HF & EF<35% significantly reduced mortality (SCD-HEFT)
- CRT, when further added to ICDs, reduces HF exacerbations by 41%. Similar benefits for ischemic and non-ischemic cardiomyopathy as well as significant reduction in LV volume & EF improvement (MADIT-CRT)
- Mechanical circulatory support (i.e. intra-aortic balloon pump, IABP)
- An IABP deflates in diastole and inflates in systole to decrease impedance to LV ejection of blood and increase coronary perfusion
- Used in extreme cases of unstable hemodynamics
- Diuretics
- Mainstay of symptomatic congestion control; may help improve stroke volume by eliminating excessive circulating volume and decreasing preload, therefore reducing “backup” of fluid into pulmonary interstitium and peripheral tissues. Diuretics have not shown mortality benefit.
Acute decompensation
Can J Cardiol. 2008 Jul;24 Suppl B:9B-14B.
CMAJ. 2007 March 13; 176(6): 797–805.
It is important to identify the precipitant of heart failure and treat as the HF episode is being treated.
- Diuretics: Lasix (furosemide)
- Helps eliminate excess fluid that the heart cannot accommodate (preload) and improve stroke volume, thereby decreasing pulmonary and peripheral edema
- Morphine:
- Decreases preload by acting as a venodilator, and reduces sympathetic activation and consequently demand on heart by procuring pain relief
- Nitrates:
- Decrease preload via venodilation, and improve oxygen delivery to the heart
- Oxygen:
- Oxygen +/- noninvasive ventilation – preserve ventilator drive and maintaining blood oxygen saturation
- Positioning:
- Sit patient upright with legs dangling down to promote blood pooling in the lower extremities and decrease preload